Синдром Фанкони: признаки и лечение. Синдром Фанкони: симптомы, диагностика и лечение При одной из форм синдрома фанкони
Синдром Фанкони (глюкозо-фосфат-аминовый диабет, болезнь де Тони-Дебре-Фанкони, первичный изолированный синдром Фанкони) - генетическое заболевание, развившееся в результате аутосомно-рецессивной мутации, характеризующееся нарушением обратного всасывания воды и биоактивных веществ из первичной мочи (канальцевая реабсорбция), обусловленное поражением почечных канальцев. Относится к рахитоподобной группе заболеваний, при которых происходят системные метаболические изменения.
Причины
Патологические изменения представляют одну из форм гиперпаратиреоза - эндокринного расстройства, развивающегося при избыточной выработке паратгормона (производится паращитовидными железами) в результате гиперплазии желез или злокачественного поражения.
Варианты наследования синдрома де Тони-Дебре-Фанкони:
- Аутосомно-доминантный - дефектный ген наследуется от одного из родителей (семейная форма).
- Аутосомно-рецессивный - дефектный ген присутствует у обоих родителей. В случае с синдромом речь идет о локальной форме аутосомно-рецессивного наследования (хромосома 15q15.3.)
Синдром Фанкони у детей может быть компонентом других генетических заболеваний:
- Цистиноз - чрезмерное накопление цистина (аминокислота) в цитоплазме (внутренняя жидкая клеточная среда) клетки.
- - нарушение преобразования галактозы (моносахарид) в глюкозу, обусловленное мутацией гена, отвечающего за выработку галактозо-1-фосфатуридилтрансферазы (фермент).
- Тирозинемия I типа - недостаток фумарилацетоацетата гидролазы, приводящей к нарушению метаболизма тирозина.
- - тяжелая гепатоцеребральная дистрофия, обусловленная нарушением метаболизма меди.
- фруктозы - потеря фермента в результате нарушения всасывания фруктозы, ее непереносимость, обусловленная дефицитом белка-транспортера фруктозы.
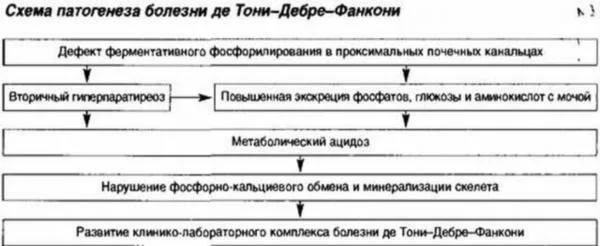
Установлено, что в основе патологии лежит комбинированная тубулопатия - группа болезней, при которых нарушен транспорт биологически активных веществ в канальцевой системе. Главное звено механизма развития синдрома Фанкони - дефект митохондрий (энергетическое депо клетки) в цикле трикарбоновых кислот (цикл Кребса), являющейся ключевой стадией дыхания клеток.
Этапы механизма развития болезни, где каждый последующий этап будет следствием предыдущего, можно представить следующим образом:
- Митохондриальный дефект, ферментная тубулопатия.
- Нарушение реабсорбции аминокислот и ферментов в канальцах почек.
- Накопление кислот ().
- Костная резорбция (разрушение).
- Нарушение обратного всасывания кальция и калия в канальцах.
Клетки теряют энергообеспечение, в результате чего развиваются тяжелые нарушения обмена веществ. Среди факторов риска выделяют:
- отравление тяжелыми металлами;
- токсикоинфекции;
- прием просроченных антибиотиков тетрациклинового ряда;
- дефицит витамина D;
- (нарушение белкового обмена).
Классификация
Выделяют первичную (идиопатическую) и вторичную формы болезни Фанкони. Первичная форма развивается в результате наследования дефектного гена. Вторичная форма возникает при других врожденных, генетически обусловленных заболеваниях.
Вторичный синдром может возникать на фоне приобретенных патологий:
- парапротеинемия - наличие в крови аномальных белковых тел;
- - тяжелые нарушения, развивающиеся при поражении клубочков почек;
- тубулоинтерстициальные - группа болезней почек, характеризующихся первичным поражением канальцев;
- злокачественное новообразование (паранеопластический синдром);
- при отравлениях;
- ожоги тяжелой степени.
Симптомы
У синдрома Фанкони, симптомы которого появляются у детей ко второму году жизни, имеется два варианта развития, в зависимости от клинико-лабораторных показателей:
- Первый вариант - характерно тяжелое течение, выраженное отставание в физическом развитии, переломы и деформации костей в результате гипокальциемии, нарушение всасывания в кишечнике.
- Второй вариант - относительно легкое течение, умеренные признаки задержки в физическом развитии, деформации костей при нормальном уровне кальция, всасывание кальция в кишечнике в пределах нормы.
Первые симптомы болезни у детей до двух лет:
- резкое снижение аппетита;
- дефицит массы тела;
- вялость;
- (нарушение пищеварения, обусловленное белково-энергетической недостаточностью);
- жажда;
- низкое артериальное давление;
- (большое количество мочи);
- субфебрильная температура;
- рвота.
Такие дети примерно к пяти годам не могут ходить.
При разгаре заболевания к пяти-шести годам первым признаком будет выступать (костное размягчение), костные деформации, параличи, связанные с недостатком кальция.
После появления первых признаков наблюдается отставание в умственном и физическом развитии. Генерализованная (распространенная на весь организм) декальцификация проявляется деформациями нижних конечностей (вальгусная деформация - искривление вовнутрь, варусная - искривление вовне), потеря мышцами тонуса, искривление грудной клетки, костей предплечий и плеч. Нехватка фосфора у детей приводит к появлению .
При прогрессировании синдрома у детей выявляются зрительные расстройства, болезни нервной системы, мочевой и пищеварительной системы, болезни ЛОР-органов. Редко возникают .
При синдроме Фанкони у взрослых происходит остеомаляция, обусловленная дефицитом минералов и микроэлементов. Пациенты жалуются на боли в костях, слабость в мышцах, вялость, возможно повышение артериального давления, развитие почечной недостаточности при отсутствии терапии.
У детей раннего возраста даже на первых неделях жизни могут возникнуть признаки синдрома Висслера-Фанкони, обусловленные выраженной аллергической реакцией. Характеризуется патология лихорадкой, эритематозной сыпью, поражением суставов (чаще рук).
Диагностика
Для выявления болезни Фанкони используют лабораторные и визуальные методы диагностики. Биохимическое исследование крови позволяет выявить недостаток кальция и фосфора, бета-2-микроглобулин (низкомелекулярный белок). В моче выявляется аминоацидурия (продукты обмена аминокислот), почечный (электролитные нарушения), гликозурия (сахар в урине), большое количество фосфатов, дефицит микроэлементов (натрия, кальция, калия, фосфора и других).
В оценке функции почек большую роль играет ультразвуковое обследование и МРТ.
Рентгенологическое исследование костей позволяет изучить костную структуру, обнаружить нарушения структуры, степень остеопороза, деформацию, оценить возрастное отставание развития костной ткани. При болезни Фанкони с помощью рентгенографии обнаруживают:
- грубоволокнистую структуру кости;
- - разрушение эпифизарной (хрящевой) пластинки роста;
- ячеистую структуру и шпороподобные наросты в большеберцовых костях;
- и переломы - на поздних стадиях.
При позитронно-эмиссионном исследовании (ПЭТ) выявляют накопление радиоизотопного вещества в зонах роста костей больного.
В биопсийном материале находят нарушение структуры кости, лакуны (патологические углубления), плохую минерализацию.
При глюкозо-фосфат-аминовом диабете проводится дифференциальная диагностика с такими патологиями:
- цистиноз;
- гликогеноз;
- разные синдромы (Лоу, нефротический);
- множественная миелома (злокачественная болезнь крови);
- непереносимость фруктозы, обусловленная генетическим фактором, другими наследственными заболеваниями;
- состояния, возникающие при трансплантации почки.
Лечение
Лечением синдрома Фанкони занимается гематолог и генетик. При , аномалиях строения почечных структур, выраженной протеинурии необходима консультация уролога и нефролога, при эндокринных расстройствах - эндокринолога, при нарушениях зрения - офтальмолога.
Терапия направлена на:
- устранение электролитных нарушений, в частности тубулярного ацидоза;
- коррекцию кислотно-щелочного дисбаланса;
- устранение клинических проявлений (симптоматическая терапия).
Курсом назначаются препараты калия и кальция, витамин D c постепенным увеличением дозировки и исследованием крови в динамике на содержание фосфора и кальция. Рекомендовано обильное питье и диета. В питании следует ограничить потребление соленых продуктов и соли, ввести в рацион молоко, курагу, чернослив, фруктовые соки. При нормализации показателей крови можно делать массаж, принимать хвойные ванны.
Хирургическое вмешательство показано только при выраженных деформациях костей. Выполняется операция при стойкой ремиссии продолжительностью от полутора лет, которая подтверждена диагностическими показателями и клиническими проявлениями.
Самое тяжелое осложнение глюкозо-фосфат-аминового диабета - . При тяжелой степени заболевания, угрожающей для жизни пациента, показан гемодиализ («искусственная почка»).

При некоторых видах почечной недостаточности гемодиализ проводится временно, до улучшения или восстановления почечной функции. В остальных случаях, при необратимых процессах в почках, процедура проводится пожизненно.
Диализ заключается в пропускании крови по специальной системе, где из биожидкости отделяются токсические вещества, которые удаляются с помощью диализирующего раствора. Организм освобождается от отравляющих продуктов распада до следующего накопления.
Прогнозы
Прогноз зависит от основного заболевания, на фоне которого развился синдром. Если основная болезнь - новообразование, при успешном его удалении прогноз может быть относительно благоприятным.
Синдром Фанкони (полное название – де Тони-Дебре-Фанкони) – врожденная патология, которая выражается в серьезной дисфункции проксимальных почечных канальцев, а именно – нарушении вторичной абсорбции (всасывании в кровь) отфильтрованных почками веществ, которое приводит к глюкозурии (повышенному сахару в моче), фосфатурии (нарушению обмена фосфора и кальция), аминоацидурии (повышенному выведению аминокислот с мочой) и уменьшению концентрации гидрокарбонатов, регулирующих кислотность крови.
Синдром Де-Тони-Дебре-Фанкони
Синдром Фанкони очень редкое заболевание, в основном встречающаяся у детей, и по медицинской статистике ее частота соответствует 1 больному малышу на 350 тысяч новорожденных обоих полов.
У взрослых людей наблюдается крайне редко, развиваясь на фоне приобретенных патологий. Код патологии согласно МКБ-10: E72.О.
Причины
Характер и причины генетического порока синдрома Фанкони сегодня изучены недостаточно.
Предположено, что в основе патологии лежат или пороки транспортных белков почечных канальцев или генная мутация, искажающая функцию ферментов, регулирующих обратное всасывание глюкозы, аминокислот и фосфора.
Имеются данные исследований о точечных дефектах митохондрий, приводящих к неправильному функционированию канальцев почек.
Болезнь также связывают с непереносимостью фруктозы, хроническим отравлением токсинами (тяжелых металлов, ифосфамида, аминогликозидов), дефицитом витамина D, амилоидозом, недостаточностью целого ряда клеточных ферментов (пируваткарбоксилазы, фосфоенолпируваткарбоксикиназы и прочих), тирозинемией, метахроматической лейкодистрофией, галактоземией, цистинозом, гликогенозами.
По мнению других специалистов, синдром Фанкони может являться изолированной патологией – а именно – одной из тяжелых форм рахитоподобных патологий, имеющих наследственный характер.
Исследования подтверждают, что при синдроме Фанкони нарушен клеточный энергетический обмен с участием АТФ (аденозинтрифосфат) и межклеточный транспорт в канальцах главного элемента почки - нефрона.
Вследствие недостаточности функций ферментов происходят потери глюкозы, фосфатов, аминокислот, и канальцы почек испытывают дефицит энергии. При этом важные вещества уходят с мочой, приводя к дистрофическим изменениям костной ткани – рахиту.
Так как в медицине пока не пришли к однозначному выводу о причинах синдрома Факони, это состояние обозначают и другими терминами: «глюкофосфаминный диабет», «идиопатический ренальный синдром Фанкони», «D-резистентный рахит», «почечный нанизм с D-резистентным рахитом», «наследственный синдром Фанкони».
Формы и патогенез
Выделяют два вида синдрома Фанкони:
- наследственный (врожденный, идиопатический), который относится к первичной форме;
- приобретенный, который рассматривают как вторичную форму болезни.
Наследственный (генетический) вид специалисты связывают с дефектом X-хромосомы, который наследуется по доминантному и рецессивному типу, поэтому генетический прогноз его проявления у будущего потомства – задача не простая. Если патология относится к врожденному (первичная форма) типу, то она выявляется у ребенка в грудном периоде до года. Поэтому первичную форму называют «младенческой».
Степень генной мутации обуславливает и степень выраженности самого синдрома. Так, наследственный полный синдром Фанкони обнаруживает себя при наличии 3 базовых биохимических дефектов, к которым относят глюкозурию, аминоацидурию, фосфатурию, неполный - при двух из них.
Обычно генетически обусловленная патология сопровождается другими врожденными недугами: цистинозом, синдромами Вильсона, Дента, Лоу, непереносимостью фруктозы, тирозинемией (неспособность организма эффективно расщеплять аминокислоту тирозин), галактоземией (дисфункция преобразования сахара в глюкозу), избыточной аккумуляцией гликогена.
Развитие синдрома Фанкони
Приобретенный синдром (вторичный), в отличие от врожденного, не сопровождается, а является следствием уже имеющихся патологий:
- тирозинемия I типа;
- цистиноз (нарушение обмена аминокислоты цистина с последующим поражением почек);
- непереносимость фруктозы;
- болезнь Вильсона-Коновалова;
- галактоземия;
- гликогеноз (аномальное скопление гликогена в тканях и органах) тип XI;
- наследственные патологии почек;
- (нарушение белкового обмена, приводящее к склерозу, атрофии, дисфункции органов);
- тубулоинтерстициальные (нефриты с поражением ткани, канальцев почек);
- гиперпаратиреоз (эндокринное заболевание, при котором нарушается нормальное содержание в крови кальция и фосфора);
- злокачественные образования: миелома, легких, поджелудочной железы, легких, болезнь легких цепей, ;
- глубокие ожоги.
Кроме того, синдром могут спровоцировать такие состояния:
- пересадка органа при низкой совместимости тканей;
- дефицит витамина D;
- отравление ураном, висмутом, ртутью, свинцом, кадмием;
- контакт с толуолом, малеиновой кислотой, лизолом;
- применение нефротоксичных фармакологических средств, таких как: Гентамицин, препараты платины, просроченные лекарства на основе тетрациклина, Диданозин, Цидофовир, медикаменты противораковой химиотерапии – Ифосфамид, Стрептозоцин.
Симптомы и признаки
При наследственной (врожденной) форме
Первичные симптомы проявляются в первые месяцы жизни, редко – после полутора лет.
Прежде всего, у новорожденного ребенка замечают следующие состояния:
- частое мочеиспускание (полиурия);
- повышенная жажда (полидипсия);
- длительные запоры;
- частые приступы беспричинной рвоты;
- астения (общая утомляемость), мышечная слабость;
- необъяснимые «скачки» температуры до 37,5 – 38 C;
- вздутый животик.
Как правило, в период, когда начинаются приступы рвоты и подъемы температуры малыша показывают педиатру. Опытный специалист должен определить, что сочетание беспокоящих родителей признаков не имеет отношения к ОРЗ, ОРВИ или энтеровирусной инфекции.
Грамотный детский врач способен вовремя распознать синдром Фанкони. А лабораторные исследования – подтвердить подозрения, если выявляют три (или два) базовых признака: глюкозурию, генерализованную гипераминоацидурию и гиперфосфатурию, характерных для этой патологии.
После слабовыраженных и достаточно неопределенных симптомов в последующие год – полтора четко фиксируются свойственные синдрому Фанкони симптомы:
- Ранний нанизм (низкорослость), вызванный постоянным выведением из организма важнейших аминокислот, глюкозы, кальция, фосфатов. Первые полгода нормального роста и веса сменяются дефицитом массы тела (до 30%) и роста (от 2 до 21%).
- Рахит, обусловленный массивным выведением кальция и фосфатов, становится заметным после 10 – 12 месяцев жизни, причем имеет характерные для синдрома Фанкони особенности: головка малыша обычно мало деформирована, но крупные кости ножек и ручек показывают существенные искривления - деформации по варусному типу, когда голени малыша искривляются «колесом», или вальгусному (в виде буквы «Х»). Искривляются и кости грудной клетки, позвоночника.
- Задержка в умственном и физическом развитии.
- Необщительность, пугливость, закомплексованность.
- Полидипсия и полиурия могут прогрессировать и регрессировать, не проходя окончательно.
- Умеренная мышечная гипотония, выражающаяся в замедленности, затрудненности движений, приводящая к тому, что дети 5 – 6 лет не способны ходить.
- Боли в костях, умеренной интенсивности, мешающие ребенку ходить. Сильнее проявляются на уровне ног, таза и позвоночного столба. Походка, если ребенок ходит, становится «утиной», неуверенной.
- Высокая вероятность переломов трубчатых костей из-за дефицита минералов в костной ткани.
- Остеомаляция или размягчение костей из-за разрушения костной ткани в результате нехватки солей кальция и фосфора.
- Сниженная иммунная защита к инфекциям, что проявляется в частых вирусных заболеваниях, отитах, пневмониях.
- Параличи, вызванные нехваткой калия.
- Офтальмологические патологии, такие как: пигментный ретинит, врожденная катаракта.
- Развитие патологий нервной системы, ЛОР (ухо, нос, глотка, гортань) и ЖКТ-органов, сердечно-сосудистой системы, анатомические аномалии органов мочевой системы вследствие массивных нарушений обмена веществ.
- В единичных случаях - эндокринные нарушения
При прогрессировании тубулярных расстройств (нарушение транспорта органических веществ, минералов, электролитов) к 10 – 12 годам у детей повышена вероятность развития хронической почечной недостаточности, что угрожает его жизни.
Видимые симптомы синдрома Фанкони у детей

У взрослых пациентов при развитии вторичной формы
Если приобретенный синдром Фанкони развивается у людей взрослых при других болезнях или патологических состояниях, то его проявления, часто сочетаются с проявлениями заболевания-провокатора.
Тем не менее, выявлены базовые признаки:
- Увеличенный объем мочи в сутки (до 2 литров и более) и острая жажда, свойственные также пациентам раннего детского возраста.
- Общая и мышечная слабость, боли в костях.
- Высокая вероятность упорного повышения кровяного давления () на фоне дисфункции почек.
- Остеомаляция (разрушение костей).
- Ацидоз (повышение кислотности крови) по причине задержки продуктов окисления в организме, приводящий к гипокалиемии (дефициту калия).
- Нефрокальциноз - высокое отложение солей кальция в почках с характерными для этого состояния лихорадкой, ознобом, тошнотой, сильнейшими болями в животе, паху, яичниках.
- Гипокалиемия (низкое поступление калия), вызывающая тяжелые сердечные осложнения, включая угрожающие жизни аритмии.
- Быстрое (при отсутствии лечения) формирование хронической недостаточности почек
У женщин
Наиболее неблагоприятный вариант течения синдрома Фанкони реализуется при его развитии у женщин в периоде постменопаузы. В это время, на фоне снижения продукции гормонов происходит естественное уменьшение плотности костной ткани (остеопения).
При сочетании этого состояния с увеличением хрупкости костей по причине нехватки минералов, высока вероятность тяжелых компрессионных переломов позвонков, шейки бедра и последующей инвалидностью.
Диагностика
Чтобы подвердить или опровергнуть диагноз, с помощью рентегографии проводят обследование костей и углубленные биохимические исследования крови и мочи.
Лабораторная
Выявляемые изменения в биохимии мочи и крови:
| Признаки | Показатели |
|---|---|
| низкое содержание кальция и фосфора | менее 2,1 ммоль/л и 0,9 ммоль/л соответственно |
| ацидоз («закисление» крови) | ВЕ = 10 – 12 ммоль/л |
| глюкозурия (увеличение сахара в моче) | 2 – 3% и выше |
| гипераминоцидурия (выведение с мочой важных аминокислот аланина, аргинина, глицина, пролита) | до 2 – 2,5 г/сутки |
| выведение кальция с мочой | 1,5 – 3,5 ммоль/сут |
| увеличение рН (кислотности) мочи из-за аномально высокой потери бикарбонатов | до 6,0 |
| увеличение относительной плотности мочи | 1,025 – 1,035 |
При обследовании также обнаруживают:
- увеличение активности щелочной фосфатазы;
- чрезмерное выведение из организма солей натрия, калия;
- протеинурию (появление белка в моче) при наличии легких цепей иммуноглобулинов, лизоцима, низкомолекулярных белков, бета 2-микроглобулинов;
- повышение клиренса (скорости фильтрации) мочевой кислоты с ее пониженным сывороточным содержанием
- падение активности ферментов энергообмена: сукцинатдегидрогеназы, а-глицерофосфатдегидрогеназы, глутаматдегидрогеназы;
- повышение в крови количества молочной и пировиноградной кислоты.
Инструментальная
Диагностирование синдрома Фанкони предусматривает обязательное использование рентгенографии костей с целью выявления деформации скелета, конечностей, обнаружения признаков остеомаляции, остеопороза, а у детей – дополнительно – задержки роста костей в сравнении с нормой по календарному возрасту.
Наблюдаются следующие аномалии в костной ткани:
- грубоволокнистая, ячеистая структура с лакунами, слабой минерализацией, аномальных разрастаний в виде «шипов» в бедренных и берцовых костях;
- признаки эпифизиолиза (частичная или полная остановка роста кости в длину в несозревшем скелете, приводящее к асимметрии конечностей);
- переломы трубчатых костей (на поздней стадии) на фоне развития остеопороза, степень которого определяют с помощью рентгеноденситометрии;
- аккумуляция радиоизотопа в областях интенсивного роста кости.
В почках:
При электронном исследовании биопсии ткани почки (биопсии) выявляют характерное изменение формы канальцев в виде «шеи лебедя», истончение, атрофию (уменьшение объема) эпителиальной ткани при наличии повышенного количества митохондрий в ней, фиброз (аномальное разрастание) соединительной ткани.
Необходимые исследования:
Дифференциальная
Патологию следует отличать от всех изолированно протекающих болезней, которые способны провоцировать возникновение приобретенного синдрома Фанкони, приобретенных нездоровых состояний и интоксикаций.
Кроме того, у грудничков педиатр обязан дифференцировать состояние острой нехватки витамина D при синдроме Фанкони от его переизбытка при употреблении искусственных добавок или расстройстве кальциевого обмена.
Различия при гипервитаминозе D и синдроме де Фанкони у детей до года:
| Параметры | Гипервитаминоз D | Синдром де Фанкони |
|---|---|---|
| Частота | Часто | Редко |
| Симптомы (схожие) | Сухость кожи, бледность, острая жажда, рвота, длительные запоры, дефицит веса и роста, увеличение печени | |
| Симптомы (различающиеся) | Гипертензия (частые подъемы кровяного давления) | Истощение, полиурия, гипотония мышц, повышенное давление крови отсутствует |
| Кровь | Избыток кальция в остром периоде. Уменьшенное содержание фосфора. Щелочная фосфатаза, сахар, белок - в норме | Кальций чаще в норме (может быть понижен). Глюкоза, белок снижены. Фосфор снижен резко. Активность щелочной фосфатазы резко повышена - в 2 – 3 раза. Кислотность крови повышена |
| Моча | Тест Сулковича на выведение кальция положительный. Наличие белка, крови (более 2 – 3 эритроцитов в поле зрения), лейкоцитов. Сахар аминоазот, как правило, в норме | Тест Сулковича отрицательный. Белок, фосфаты, сахар повышены, аминоацидурия |
| Кости | Зоны обызвествления расширены, уплотнены | Остеопороз трубчатых костей, дефицит кальция в областях обызвествления |
Специалисты, занимающиеся синдромом Фанкони и его осложнениями: нефролог, ортопед, гематолог, эндокринолог, уролог, офтальмолог, генетик.
Лечение
Рациональная, хорошо продуманная терапия способна уменьшить влияние на мозг, костную систему и органы чрезмерных потерь важнейших аминокислот, минералов, глюкозы и белка, выводимых в составе мочи.
Поэтому лечение при синдроме нацелено на следующие задачи:
- Максимально возможная коррекция дефицита калия, бикарбонатов, изменений в кислотно-щелочном балансе крови для уменьшения ацидоза.
- Терапия фосфат-диабета (D-резистентного рахита) с акцентом на недопустимости ограничения жидкостей.
- Лечение основного заболевания, провоцирующего развитие приобретенного синдрома у взрослых.
Медикаментозное
 Для смягчения потерь фосфора и кальция используют специальные препараты с витамином D, это l,25(OH)D3 и l(OH)D3.
Для смягчения потерь фосфора и кальция используют специальные препараты с витамином D, это l,25(OH)D3 и l(OH)D3.
Начальные дозы витамина D3 в сутки 10 – 15 тыс. ME. Повышение дозы проводят постепенно, увеличивая ее каждые 12 – 14 дней (под контролем пробы Сулковича и содержания в крови фосфора). При отсутствии признаков интоксикации и небольшом выведении кальция с мочой разрешается увеличивать дозу, доводя до 100 – 150 тыс. ME в сутки, и продолжать терапию до нормальных показателей в крови фосфора и щелочной фосфатазы. При стабилизации их значений повышать дозу далее не следует.
Терапия витамином D проводится несколькими курсами с целью предупреждения кризов прогрессирования рахитических деформаций в костях.
Оптимальным вариантом считают использование активных метаболитов D3 – Оксидевит (0,5 – 1,5 мкг в день), кальциотриол (Рокальтрол).
Включают препараты кальция (кальция глюконат в сутки до 1,5 – 2 грамм), фосфора (0,5 – 1 грамма в сутки), фитина.
Из неорганических фосфатов используют смесь Олбрайта, принимая его по 1 большой ложке 4 – 5 раз в сутки. Применяют фосфаты в виде раствора и таблеток в дозах, рассчитанных по 10 мг на килограмм веса, 4 раза в день (обязательно с препаратами витамина D во избежание гиперпаратиреоза).
При выраженном дефиците калия применяют Панангин, Аспаркам.
При любых назначениях постоянно отслеживают КОС или кислотно-основное состояние крови. В норме кровь имеет слабощелочную реакцию, а рН в диапазоне 7,35 – 7,45. При ацидозе, когда величина рН снижается ниже 7,35, кровь приобретает повышенную кислотность.
В этих случаях показано внутривенное вливание раствора натрия гидрокарбоната 4% или питье раствора (50 – 60 мл в день), в который входит лимонная кислота — 2 грамма, цитрат натрия – 3 г, цитрат калия – 3,3 г, вода 100 мл. В 1 мл такой ощелачивающей микстуры содержится по 1 ммоль натрия и калия. Высокая кислотность крови нейтрализуется также с помощью питьевой соды (бикарбоната натрия).
Некоторые специалисты на основе практических результатов рекомендуют Унитиол как средство, повышающее активность тиолзависимых ферментов.
При цистинозе назначают: Дитиотрентал из расчета 25 мг на килограмм веса пациента через 3 часа; Цистеамин в суточной дозе из расчета 90 мг/кг.
Имеются положительные результаты применения Пеницилламина, понижающего в крови концентрацию пировиноградной кислоты, уменьшающего степень выведения аминокислот и способствующего росту в организме резервов щелочи.
Хорошее влияние оказывают на работу канальцев почек гормональные анаболики, включая Метилтестостерон.
Лечение в стационаре показано при явно выраженных обменных нарушениях, включая гипо- и гипергликемию, деформациях скелета.
Профилактика
Современная профилактика врожденного синдрома Фанкони при наличии подобной патологии в семье заключается в предварительном генетическом консультировании. Риск развития болезни для так называемых сибсов, то есть, сестер и братьев - около 25%.
При вторичной форме синдрома его симптомы уменьшаются или полностью исчезают при активном лечении основной болезни или приобретенного патологического состояния.
Прогноз
Прогноз заболевания зависит от формы (первичная, вторичная) синдрома, тяжести проявлений, и начала лечения.
Например, симптоматика приобретенного синдрома исчезает при устранении причины-провокатора.
При врожденной патологии на фоне отсутствия отложений цистина в тканях течение болезни не несет серьезной угрозы жизни, в ином случае, особенно, без соответствующего лечения, гибель пациента прогнозируется до 10 – 20 лет от нарастающей недостаточности почек.
Однако, даже при тяжелых изменениях в почках: пиелонефрите, тубулоинтерстициальном нефрите, почечной недостаточности, при раннем медикаментозном воздействии на патологический процесс и длительной терапии устанавливается определенное равновесие в гомеостазе, при котором прогноз нормального качества жизни на десятки лет – хороший.
В медицинской практике встречаются истории болезни, когда у детей 7 – 8 лет наследственный синдром Фанкони практически «купировался» с наступлением длительной ремиссии, явного улучшения состояния ребенка и даже – выздоровления.
Синдром Фанкони представляет собой системное нарушение обмена веществ, при котором выявляется дисфункция почек. Они утрачивают способность обратного всасывания и возвращения в кровоток аминокислот, глюкозы, солей кальция и фосфора, выводящихся с мочой. Это врожденный или приобретенный органический дефект. Встречается он чрезвычайно редко, 1 случай приходится на 350 тысяч новорожденных. Однако эта дисфункция крайне тяжело отражается на состоянии организма.
Ее называют также синдромом де-Тони-Дебре-Фанкони, фосфат-диабетом, наследственным синдромом Фанкони. Одни ученые полагают, что эта патология возникает, когда в клетках резко уменьшаются запасы АТФ (аденозинтрифосфорной кислоты). Другие выдвигают версию о том, что рахитичная деформация костей происходит из-за дефицита фосфора, сбоя в кислотно-щелочном балансе либо из-за обоих этих факторов. Третьи считают, что дисфункция почек обусловлена не биохимическим, а структурным дефектом. Точная же причина до сих пор не установлена.
При такой патологии поражаются почечные проксимальные канальцы. Концентрация соединений кальция и калия в кровотоке сохраняется в пределах нормы. Реакция мочи - щелочная или нейтральная. Из-за значительных потерь гидрокарбонатов развивается ацидоз. Это опасное состояние, при котором происходит нарушение кислотно-щелочного баланса и «закисление» организма. Ацидоз, как правило, вызывает любая диффузная болезнь почек.
Врожденная патология может иметь различные степени тяжести. При наличии 3 биохимических дефектов диагностируют полную, а при 2 — неполную дисфункцию почек. Некоторые ученые считают, что наследственный синдром Фанкони - самостоятельное рахитоподобное заболевание. Однако чаще всего оно сопутствует другим врожденным патологиям. Это могут быть:
- цистиноз (заболевание, связанное с кристаллизацией аминокислоты цистин в тканях);
- болезнь Вильсона-Коновалова (патология, вызываемая отложениями меди в почках, печени, мозге);
- галактоземия (нарушение преобразования простого сахара в глюкозу);
- синдром Лоу (патология, проявляющаяся задержкой роста, психического развития, катарактой и глаукомой);
- тирозинемия (пассивность фермента печени, расщепляющего аминокислоту тирозин);
- непереносимость фруктозы.
Приобретенная дисфункция, названная именем Фанкони, может возникнуть из-за таких факторов:
- медикаментов, токсичных для почек (Аспирина, Гентамицина, просроченного Тетрациклина, антивирусных средств Цидофовир, Диданозин, противоопухолевых препаратов Стрептозоцин, Ифосфамид и др.);
- отравления солями тяжелых металлов, другими агрессивными химическими соединениями;
- острого дефицита витамина D;
- множественной миеломы (рака крови);
- амилоидоза (нарушения белкового обмена);
- трансплантации почки.
Симптомы болезни у детей
Наиболее ярко проявляется врожденный синдром Фанкони у детей. Его симптомы дают о себе знать уже на первом году жизни малышей. Это:
- полиурия (учащенное мочеиспускание с большим количеством выводящейся из организма жидкости);
- полидипсия (патологически сильная жажда);
- повышенная температура;
- судороги;
- частая рвота без видимых причин;
- затяжные запоры;
- кожная сыпь;
- отечность суставов;
- увеличение почек, лимфоузлов, селезенки;
- предрасположенность к инфекциям.

Из-за ежедневного выведения из организма с жидкостью витаминов, микроэлементов, глюкозы ребенок отстает в умственном и физическом развитии. У него искривляются кости ног, становятся дряблыми, а потом и вовсе атрофируются мышцы. Нередко дети утрачивают способность ходить самостоятельно. К 12–13 годам развивается хроническая почечная недостаточность. Иногда ухудшается зрение. Кроме того, при синдроме Фанкони симптомы патологии могут свидетельствовать о сочетанных врожденных дефектах сосудов и сердца, органов пищеварения, мочеполовой системы.
Симптомы болезни у взрослых
Чаще всего отмечаются:
- полиурия;
- гипостенурия (снижение относительной плотности мочи);
- ломота в костях;
- остеомаляция (размягчение костных тканей и утрата ими прочности);
- мышечная слабость;
- ускоренное прогрессирование гипертонии;
- хроническая почечная недостаточность (если отсутствует лечение).
Полиурия при синдроме Фанкони у взрослых не выражена резко, этим она существенно отличается от избыточного количества мочи при несахарном диабете. Чаще всего данный синдром становится проявлением множественной миеломы или болезни Вальденстрема (злокачественного поражения кроветворной системы). Кроме умеренной полиурии, нарушение деятельности почек проявляется снижением их концентрационной функции, появлением белка в моче.
О том, есть ли синдром Фанкони или нет, можно судить по таким характерным симптомам, как высокая концентрация в моче аминокислот, глюкозы и фосфатов. Повышенное выведение глюкозы с мочой при этой патологии обусловлено дисфункцией почек. Однако концентрация сахара натощак и уровень гипергликемии у этих больных, как правило, в пределах нормы.
Полиурия всегда сопровождается мышечной слабостью, которая острее всего ощущается в конечностях. Возникает она из-за дефицита калия. Кроме того, практически всегда больных донимает ломота в костях.

Симптомы ацидоза у взрослых и детей одинаковы. Это:
- потеря аппетита;
- беспричинная рвота;
- запоры или поносы;
- кислый запах, исходящий от кожи или изо рта;
- снижение давления;
- головные боли;
- одышка;
- бессонница;
- упадок сил.
Тем не менее врачи отличают взрослый ацидоз от детского. Считается, что у младенцев это всегда врожденная патология. У взрослых же она может быть как дальнейшим развитием детской болезни, так и приобретенным осложнением вследствие поражения почек. Симптомы ацидоза нередко сочетаются с признаками интерстициального нефрита и мочекаменной болезни. Конкременты в почках образуются из-за больших потерь кальция с мочой. В тяжелых случаях выявляются симптомы остеопороза и остеомаляции.
Диагностика патологии
Выявление синдрома де-Тони-Дебре-Фанкони начинается со сбора анамнеза, осмотра пациента и лабораторных анализов. Характерные признаки патологии в биохимической формуле крови - низкое содержание кальция, фосфора, калия и натрия, а также глюкозы, аланина, глицина, глютаминовой кислоты. Зато в моче - высокий уровень фосфора и глюкозы, присутствуют белок и лейкоциты.
При значительной потере гидрокарбонатов с мочой и чрезмерном накоплении кислот в организме диагностируется метаболический ацидоз. Практически у всех пациентов наблюдается превышение уровня пировиноградной и молочной кислот в крови. Выявляется и падение активности ферментов энергетического обмена.
При инструментальной диагностике обязательно проводят УЗИ почек и мочеточников. Нефробиопсия (исследование миниатюрных образцов почек) позволяет увидеть деформацию проксимальных канальцев, которые вытянуты наподобие лебединой шеи. На поздних стадиях заболевания выявляется атрофия почечных клубочков.

Рентгенография деформированных нижних конечностей помогает обнаружить перерождение костных тканей. У детей нередко встречаются скрытые переломы эпифизарных хрящей, из-за которых кости впоследствии перестают расти в длину и становятся асимметричными. В большеберцовых костях выявляются новообразования, напоминающие шпоры.
На поздних стадиях развития синдрома диагностируется остеопороз. Велик риск переломов трубчатых костей. Стадию остеопороза определяют методом рентгеноденситометрии. Низкая минерализация костных тканей выявляется при исследовании их образцов, получаемых тоже с помощью биопсии.
Главные критерии диагностики:
- Значительный дефицит веса и роста ребенка.
- Слабость статико-моторных функций.
- Рахитоподобные деформации скелета (деградация структуры костных тканей подтверждается рентгенологически).
- Электролитные нарушения.
При синдроме Фанкони у взрослых врачи обязательно проводят дифференциальную диагностику, чтобы исключить схожие с ним патологии. Это:
- наследственные заболевания (цистиноз, галактоземия, болезнь Вильсона-Коновалова, тирозинемия и др.);
- хронический пиелонефрит;
- сахарный диабет;
- вторичный гиперпаратиреоз;
- множественная миелома;
- отравления лекарственными препаратами, агрессивными химическими веществами;
- обширные ожоги.
Лечение патологии
Идеально, когда синдром Фанкони лечат генетик, гематолог, но такие специалисты есть далеко не в каждом медучреждении. Чаще всего этой патологией занимаются нефролог или уролог. Если наблюдаются признаки гиперпаратиреоза (гиперсекреции паращитовидных желез), требуется консультация эндокринолога. При ухудшении зрения необходимо обследоваться у офтальмолога.

Тактикой лечения предусматриваются:
- Восполнение дефицита электролитов.
- Устранение нарушений кислотно-щелочного баланса.
- Терапия почечной недостаточности.
- Снятие болезненной симптоматики.
При медикаментозном лечении применяются:
- Кальцитриол, Оксидевит и другие препараты витамина D;
- кальция глюконат;
- Фитин, кальция глицерофосфат, алюминия фосфат;
- ощелачивающие растворы Бицитра, Полицитра;
- Индометацин, Метилтестостерон, Гипотиазид (при тяжелом поражении проксимальных канальцев);
- Панангин, Аспаркам;
- Цистин, Меркаптамин;
- антибиотики;
- кортикостероиды и др.
Поскольку синдром Фанкони имеет хронический характер, лечение проводится длительное время, большими курсами после обязательных перерывов. Зачастую удается приблизить к норме обмен веществ, снять остроту проявления недуга и предотвратить опасные осложнения. Но полностью избавиться от синдрома де-Тони-Дебре-Фанкони чрезвычайно сложно, он часто дает рецидивы.
Терапия синдрома направлена на устранение ацидоза, восполнение запасов калия, бикарбоната кальция, фосфатов и других электролитов. Важнейшую роль при этом играет лечебная диета. Необходимо обильное питье и сокращение количества соли. Принимать пищу следует малыми порциями, но часто. Углеводы нужно строго дозировать, чтобы не было избытка глюкозы в кровотоке.
При нарушении почечной функции, отвечающей за обратное всасывание полезных веществ, обнаруживается синдром Фанкони. Второе название состояния - синдром де Тони-Дебре-Фанкони. При патологии наблюдается глюкоза, белок с , происходят метаболические нарушения. Недуг передается по наследству. Диагноз ставится по клиническим результатам. Цели лечения - возмещение дефицитных состояний, устранение почечной недостаточности.
У детей сидром де Тони-Дебре-Фанкони вызывает рахит, физическое отставание, ослабленности мышечной ткани.
Описание и причины
Недуг де Тони-Дебре-Фанкони проявляется выраженной канальцевой дисфункцией, в результате которой расстраивается процесс обратного поглощения полезных для организма веществ и ионов. Одновременно повышается выделение бикарбонатов, обнаруживаются общие метаболические сбои. Особенности:
- падение содержания кальция с фосфором в кровяной плазме;
- сбой в кислотно-основном балансе, что проявляется низкой кислотностью и бикарбонатов в крови;
- нормальное выделение кальция в виде конечного метаболита с мочой при повышенном показатели естественного очищения организма от фосфатов;
- патологически высокая активность щелочной фосфатазы;
- появление сахара в моче;
- общее нарушение выработки и выведения продуктов синтеза аминокислот;
- увеличенные объемы выводимой мочи с пониженной плотностью.
 Недуг де Тони-Дебре-Фанкони у детей.
Недуг де Тони-Дебре-Фанкони у детей.
Синдром Фанкони развивается и у детей, и у взрослых. Различают врожденный и приобретенный типы патологии. По степени тяжести симптомов, а также по типу и выраженности метаболических расстройств различают 2 формы болезни:
- Первый тип проявляется в виде выраженного отставания в физическом развитии с явной деформацией костей скелета, тяжелой клинической картиной, частыми переломами на фоне резкой недостачи кальция вследствие слабого всасывания вещества в кишечнике.
- Второй вариант проявляется в виде средней степени задержки физического развития, облегченным проявлением симптомов со слабой костной деструктцией, достаточным уровнем кальция.
Причины болезни
Провоцирующие факторы делятся на те, что вызывают приобретенную форму синдрома де Тони-Дебре-Фанкони, и те, что отвечают за появление врожденной патологии. Провокаторами вторичного нарушения признаны такие состояния:
- непереносимость фруктозы;
- нехватка клеточных ферментов;
- хроническое отравление токсинами;
- острый дефицит витамина D.
Самостоятельно синдром вызван одной плохой наследственностью или свежей мутацией гена, отвечающего за метаболические процессы. Синдром де Тони-Дебре-Фанкони зачастую развивается не один, а совместно с цистинозом, галактоземией, синдромом Вильсона, первичной тирозинемией, непереносимостью фруктозы. Чаще эта форма фиксируется у детей, нежели у взрослых.
Провоцирующие факторы
 Острый дефицит витамина D может спровоцировать болезнь.
Острый дефицит витамина D может спровоцировать болезнь.
Синдром Фанкони с большей вероятностью появляется у людей со следующими наследственными нарушениями:
- сбой в процессах обмена цистина (аминокислоты в составе белков);
- непереносимость молочной продукции (даже грудного молока);
- дефекты различных ферментов, отвечающих за синтез и распад гликогена;
- сбой в обмене ароматических аминокислот;
- непереносимость фруктозы;
- проблемы с метаболизмом меди (болезнь Коновалова-Вильсона);
- нарушение обмена миелина и дисфункция фермента сульфатазы;
- постоянное воздействие токсинов на организм (лекарственных, ядов, тяжелых металлов);
- сбой в обменных процессах с участием белка, что приводит к отложению специфичного комплекса - амилоида;
- острый дефицит витамина D.
Врожденная и приобретенная
Синдром Фанкони - тяжелая рахитоподобная болезнь, которая может быть первичной (врожденной) и вторичной (приобретенной).
Врожденная патология де Тони-Дебре-Фанкони - наследственная, то есть передающаяся от родителей потомкам. Зачастую развивается совместно с другими генетическими нарушениями. Для состояния характерно острое нарушение метаболических процессов, в результате которых, наряду с дисфункцией почечных канальцев, развиваются слепота, синдром увеличенной печени, синдром со стойким снижением концентрации и тканевой активности гормонов щитовидки. Механизм наследования синдрома определяется типом патологии, совместно с которой он развивается.
Приобретенный синдром де Тони-Дебре-Фанкони зачастую провоцируется различными лекарствами:
- используемыми при лечении рака химиопрепаратами;
- антиретровирусными медикаментами;
- устаревшим тетрациклином.
Провокаторами развития вторичных форм патологии выступают осложненная трансплантация почки, онкологические заболевания крови, выраженные дефицитные состояния (преимущественно при нехватке витамина D). Если врожденное заболевание развивается еще внутриутробно и фиксируется в основном у детей, то вторичная форма может развиться и у взрослых.
Механизм развития и течения
 Нарушается обратное всасывание веществ в почках.
Нарушается обратное всасывание веществ в почках.
Синдром де Тони-Дебре-Фанкони вызван врожденной мутацией гена, отвечающего за метаболические процессы, но отмечаются случаи свежей трансформации, вызванной другими патологиями. Вследствие нарушения всасывания множества полезных веществ и ионов развиваются тяжелые дефицитные состояния:
- при нехватке аминокислот появляется выраженная дистрофия, замедляется рост и физическое развитие;
- при выведении фосфора и бикарбонатов - сбой процесса минерализации с началом разрушения костной ткани;
- при скоплении сахара в моче - нарушение регуляции углеводного обмена;
- при выведении с мочой калия - мышечная атрофия, падение АД до 80 мм РТ. ст. и ниже;
- при масштабных обменных нарушениях - деструкция почечной ткани (сужение просвета с уплощением эпителия и укорочением канальцев, в которых происходит обратное всасывание).
Симптомы у детей и взрослых
Первые проявления врожденного синдрома де Тони-Дебре-Фанкони появляются в первый год жизни, реже - с 1,5 лет. Малыш часто мочится, наблюдается субфибралитет (до 37-38°С), запор, рвота. К 6-ти годам при отсутствии лечения детки не ходят, а к 12-ти отказывают почки, что чревато смертью. Больные - необщительные, закомплексованные, пугливые. Умственная и мыслительная функция в норме. Последствия синдрома - проблемы с НС и зрением, развитие хронического иммунодефицита, дисфункция мочеполовых органов и ЖКТ.
Симптомы у детей развиваются по большей части в связи с дефицитом фосфатов (по типу фосфат-диабета) и проявляются:
- шаткостью походки;
- низким ростом;
- малой подвижностью;
- округленностью голеней;
- искривлением костей скелета (в частности, позвоночника);
- костными болями, отчего малыш не хочет ходить;
- резкое увеличение объемов выделяемой мочи;
- падение удельного веса на фоне качественного изменения состава мочи;
- мышечная атония;
- костные боли;
- гипертоническая болезнь;
- хроническая почечная недостаточность (при отсутствии лечения);
- остеопороз, остеопения на фоне падения плотности костной ткани.
Добавляю еще информации по данному синдрому.
=====================
Болезнь Дебре — Де Тони — Фанкони
Болезнь Дебре-де Тони-Фанкоии — наследственное заболевание, передающееся по аутосомно-рецессивному типу. В основе патогенеза его лежит нарушение канальциевого транспорта аминокислот, глюкозы, неорганических фосфатов и гидрокарбонатов. Снижена канальциевая реабсорбция натрия, калия, воды, что обусловлено генетически детерминированной дисплазией нефронов. Тяжесть состояния определяется поражением не только тубулярного аппарата, но и клубочков почек. Нарушение реабсорбции воды сопровождается полиурией, полидипсией, гипостенурией и рецидивами лихорадки без видимых причин. Заболевание обычно начинается на первом году жизни. На втором году жизни у ребенка выявляются отставание в физическом развитии и костные деформации грудной клетки, костей предплечий, плеча, голеней, таза.
При рентгенографии костей обнаруживаются выраженные деформации конечностей, нарушение структуры костной ткани (системный остеопороз различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от календарного возраста ребенка). Характерными особенностями болезни Дебре- де Тони -Фанкони являются понижение концентрации кальция в крови ниже 2,21 ммоль/л и фосфора до 0,9 ммоль/л, повышение активности щелочной фосфатазы в крови, метаболический ацидоз. Экскреция кальция с мочой остается нормальной (1,5-3,5 ммоль/сут), а клиренс фосфора мочи обычно повышен. Одновременно выявляется глюкозурия (2-3% и выше), генерализованная аминоацидурия (до 2-2,5 г/сут), полиурия - до 2 л/сут и более, повышение рН мочи до 6,0. Относительная плотность мочи может быть высокой (1,025-1,035). При экскреторной урографии могут находить изменения контуров почек, повышение их подвижности, наличие аномалии сосудов.
Выделяют два варианта болезни Дебре-де Тони-Фанкони.
1-й — характеризуется грубой задержкой физического развития (дефицит длины тела более 20%), тяжелым течением заболевания, выраженными деформациями костей и нередко переломами костей, выраженной гипокальциемией (1,6-1,8 ммоль/л), снижением всасывания кальция в кишечнике (менее 20%).
2-й — характеризуется умеренной задержкой физического развития (дефицит длины тела менее 13%), легким течением заболевания, умеренными деформациями костей, нормальной концентрацией кальция в крови.
Лечение. Для ликвидации нарушений фосфорно-калыщевого обмена применяется витамин D. Начинают лечение с 10 000 ME, постепенно повышая дозу до 25 000-30 000 ME, а затем до 75 000- 150 000 ME. Повышение начальных доз проводится осторожно, постепенно каждые 2 нед, до нормализации концентрации кальция и фосфора в крови. Из активных метаболитов витамина D можно использовать оксидевит в дозе 0,5-1,5 мкг/сут. В комплекс лечения включают также препараты кальция (кальция глюконат, 1,5-2 г/сут) и фосфора (0,5-1 г/сут). Лечение витамином D должно проводиться повторными курсами, так как при отмене препарата часто наступают кризы метаболического прогрессирования остеопороза и рахитических изменений в костной систем. Диета должна быть с ограничением поваренной соли и включением пищевых продуктов, оказывающих ощелачивающее действие (молоко, молочные продукты), фруктовых соков, богатых калием сухофруктов (чернослив, курага, изюм и др.), витаминов А, В, С, Е. Показаны также соляно-хвойные ванны.
=========================
Синдром Фанкони (де Тони-Дебре-Фанкони) рассматривают как «большую» канальцевую дисфункцию, характеризующуюся нарушением реабсорбции большинства веществ и ионов (аминоацидурией, глюкозурией, гиперфосфатурией, увеличением экскреции бикарбоната) и системными метаболическими изменениями.
Синдром Фанкони включает множественные дефекты реабсорбции в проксимальных почечных канальцах, что ведет к глюкозурии, фосфатурии, генерализованной аминоацидурии и уменьшению концентрации гидрокарбонатов. Симптомы у детей включают гипотрофию, задержку физического развития и рахит, симптомы у взрослых — остеомаляцию и мышечную слабость. Диагноз основывается на выявлении глюкозурии, фосфатурии и аминоацидурии. Лечение включает возмещение дефицита гидрокарбонатов, а также лечение почечной недостаточности.

